
What Are Class I, II, and III Medical Devices?
What Are Class I, II, and III Medical Devices?
Introduction
Every medical device sold in the United States — from a tongue depressor to an implantable pacemaker — must be assigned a regulatory classification before it can reach a patient. This classification is not a bureaucratic formality. It is the foundation of the entire U.S. Food and Drug Administration (FDA) oversight framework, and similar risk-tiering logic underpins device regulations worldwide, including the EU Medical Device Regulation (MDR) and ISO 13485-based quality systems.
Understanding what are Class I, II, and III medical devices is the first step for any importer, manufacturer, or healthcare institution trying to bring a product to market — or simply trying to understand the compliance burden attached to the devices they already use. Misclassifying a device, or underestimating the controls it requires, is one of the most common (and costly) mistakes in medical device compliance. This guide breaks down the class 1 2 3 medical devices framework in plain language, with the regulatory pathways, quality requirements, and real-world examples you need.
Suggested Infographic Structure
Before unpacking the details, it’s worth visualizing this system. Here’s a logical structure you (or a design team) could use to build an infographic around this article:
- Header / Hook: “Which FDA Class Does Your Device Fall Into?” — paired with a simple risk gradient bar running Low → Moderate → High.
- Three-Panel Layout: One panel per class (Class I, Class II, Class III), arranged left-to-right along the risk gradient. Each panel shows a risk-level icon, the regulatory pathway badge (General Controls / 510(k) / PMA), 2–3 representative device icons (e.g., bandage → infusion pump → pacemaker), and one key control requirement in a single line.
- Comparison Strip beneath the panels: a slim horizontal bar comparing submission type, quality system scope, and typical time-to-market across all three classes at a glance.
- “Where’s the Line?” Callout: a small side panel specifically addressing the boundary between class 1 and class 2 medical devices, since that’s where most classification confusion happens.
- CTA Footer: “Not sure which class your device falls into? Talk to a regulatory expert.”
This flow mirrors the article’s own logic — risk level → regulatory pathway → controls required — making it easy to adapt into a single scrollable graphic or a social media carousel.
How Medical Device Classification Works
The FDA assigns each device to one of three classes based on the level of risk it poses to the patient and the degree of regulatory control necessary to provide a “reasonable assurance of safety and effectiveness.” Risk here isn’t just about physical harm — it also accounts for how invasive the device is, whether it sustains life, and how much clinical judgment is required to use it safely.
Broadly speaking:
- Class I devices carry the lowest risk and require the least regulatory oversight.
- Class II devices carry moderate risk and require demonstrated equivalence to an existing device.
- Class III devices carry the highest risk, typically supporting or sustaining life, and require the most rigorous premarket review.
Core Comparison Table: Class I vs Class II vs Class III
| Criteria | Class I | Class II | Class III |
| Risk Level | Low | Moderate | High |
| Regulatory Pathway | General Controls; most exempt from premarket submission | 510(k) Premarket Notification (Substantial Equivalence) | Premarket Approval (PMA) |
| Quality System Requirement | Basic Quality System Regulation (QSR) compliance; many exempt from full QSR | Full Quality System Regulation (21 CFR 820) compliance | Full QSR plus rigorous design controls and clinical data |
| Clinical Data Requirement | Rarely required | Sometimes required (performance/bench data usually sufficient) | Almost always required (clinical trials) |
| % of All Devices | ~47% | ~43% | ~10% |
| Examples | Bandages, tongue depressors, hand-held surgical instruments | Infusion pumps, powered wheelchairs, surgical drapes | Pacemakers, heart valves, implantable defibrillators |
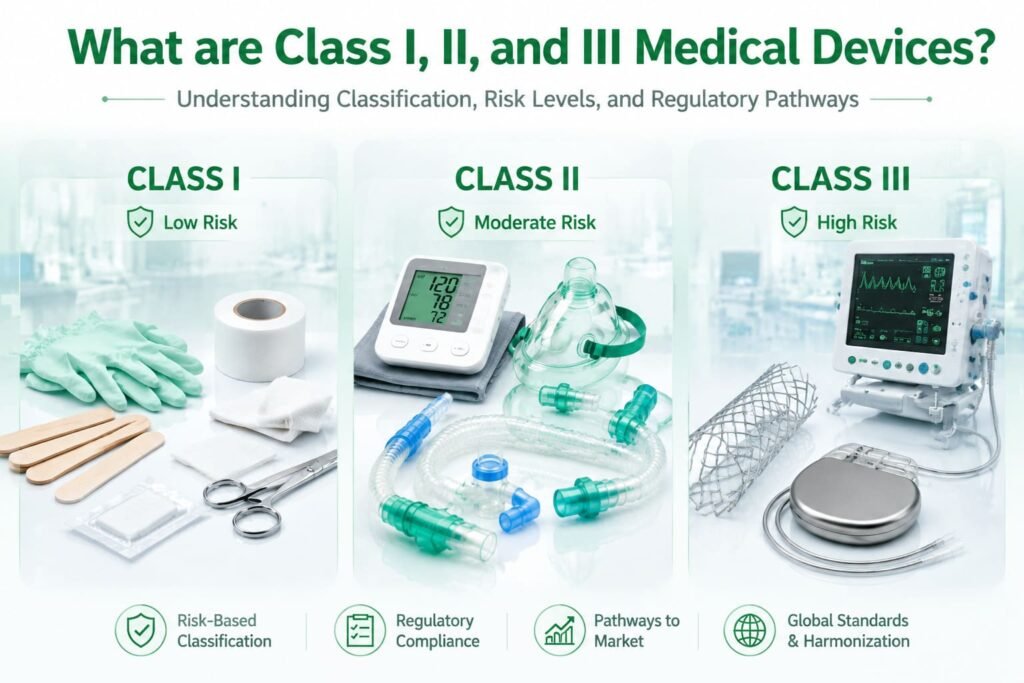
Class I Medical Devices: Low Risk, Minimal Controls
What Is a Class 1 Medical Device?
Class 1 medical devices are those posing the lowest potential risk to users. They are typically simple in design, non-invasive, and don’t sustain or support life.
Most Class I devices are exempt from the premarket notification (510(k)) process altogether, relying instead on General Controls — requirements around labeling, registration, listing, and adherence to Good Manufacturing Practice principles. However, “low regulatory burden” does not mean “no regulatory burden”: manufacturers must still register their establishment, list their devices with the FDA, and follow applicable labeling requirements.
If you’re researching medical device class 1 examples in depth for a regulatory submission or import checklist, it can help to consult a detailed list of class 1 medical devices rather than relying on a short summary. That said, the most commonly cited Class I examples include:
- Elastic bandages and adhesive dressings
- Manual toothbrushes and dental floss
- Handheld surgical instruments (scalpels, forceps)
- Examination gloves
- Tongue depressors and reusable stethoscopes
Class II Medical Devices: Moderate Risk, Substantial Equivalence
What Is a Class II Medical Device?
A Class II device is one where general controls alone are insufficient to provide reasonable assurance of safety and effectiveness, so special controls are layered on top. These may include performance standards, post-market surveillance, specific labeling requirements, and patient registries. In regulatory literature, class ii devices are often described as the “middle tier” — more heavily scrutinized than Class I, but not subject to the full clinical-trial burden of Class III.
The primary regulatory pathway for a Class 2 device is the 510(k) Premarket Notification. Rather than proving a device is safe and effective from scratch, manufacturers demonstrate that their device is “substantially equivalent” to a legally marketed predicate device already on the market. This is generally faster and less expensive than the PMA pathway required for Class III devices, but it still demands robust technical documentation, risk analysis, and often bench or biocompatibility testing.
Class 2 medical device examples commonly cited in industry include:
- Infusion pumps
- Powered wheelchairs
- Surgical drapes and gowns
- Blood pressure monitors
- Contact lenses
- Pregnancy test kits
- Absorbable sutures
- Bone plates and screws (many orthopedic fixation devices)
- Ultrasound diagnostic equipment
- Catheters (many types)
Because Class II is the largest and most diverse category, it captures the majority of devices where import compliance questions arise — making it the category importers and manufacturers most frequently need guidance on.
Class III Medical Devices: High Risk, Rigorous Oversight
Class III devices usually sustain or support life, are implanted, or present a potentially unreasonable risk of illness or injury. Because the consequences of failure can be severe, these devices are subject to the most demanding pathway: Premarket Approval (PMA).
Unlike the 510(k) route, a PMA submission requires the manufacturer to independently demonstrate safety and effectiveness — typically through controlled clinical trials — rather than relying on comparison to a predicate device. The review process is significantly longer, more expensive, and requires ongoing post-approval studies in many cases.
Typical Class III examples include:
- Implantable pacemakers and defibrillators
- Replacement heart valves
- Implanted cerebella stimulators
- High-frequency ventilators
- Certain breast implants
Class 1 vs Class 2 Medical Device: What’s the Real Difference?
When importers and manufacturers weigh class 1 and class 2 medical devices side by side, the comparison is where most compliance confusion happens — the class 1 vs class 2 medical device question comes up constantly, because on the surface the products can look similarly “low-tech.” The real distinction lies in the degree of control, not necessarily the complexity of the device itself.
Key differences:
- Premarket Submission — Most Class I devices need no premarket submission at all; most Class II devices require a 510(k) demonstrating substantial equivalence.
- Special Controls — Class II devices are subject to special controls (specific performance standards, guidance documents, or mandatory post-market surveillance) that Class I devices generally do not require.
- Quality System Scope — Class I manufacturers may qualify for exemptions from certain Quality System Regulation provisions; Class II manufacturers must generally implement the full QSR, including design controls under 21 CFR 820.30.
- Risk Profile — Class I risk is typically limited to minor, reversible harm. Class II risk includes scenarios where device malfunction could cause moderate injury, meaning tighter design verification and validation are expected.
- Time to Market — Class I devices can often reach market in weeks; Class II devices typically require several months for 510(k) review and clearance.
Understanding this gap matters most at the import and product-launch stage, where misjudging a device’s class can lead to shipment holds, warning letters, or costly redesigns.
.
Conclusion: Why Getting Classification Right Matters
Correctly identifying whether a product is Class I, II, or III is the single most consequential decision in a device’s regulatory journey. It determines your submission pathway, your quality system obligations, your time-to-market, and your exposure to enforcement risk. Get it wrong, and you risk shipment delays, FDA warning letters, or a product recall long after launch.
Given how much rides on this determination — and how nuanced predicate comparisons, special controls, and PMA requirements can be — it’s worth having your classification reviewed by a qualified regulatory affairs team before you finalize a submission or import strategy. If you’re unsure where your device falls, or need help navigating a 510(k) or PMA pathway, consulting an experienced medical device regulatory consultant can save significant time, cost, and risk down the line.
A: The core difference is the level of regulatory control required, not necessarily the device’s complexity. Most Class I devices only need to comply with General Controls and are exempt from premarket submission, while most Class II devices require a 510(k) submission demonstrating substantial equivalence to a predicate device, plus special controls like performance standards or post-market surveillance.
A: The large majority do, but there are exceptions. Some Class II devices are specifically exempted from 510(k) requirements by FDA regulation, though they still must comply with applicable special controls and general controls, including registration and listing.
A: Yes. The FDA periodically reclassifies devices — usually downward, from Class III to Class II, as more clinical evidence accumulates and risk profiles become better understood, though reclassification upward is also possible if new safety concerns emerge. Manufacturers should monitor FDA classification updates relevant to their product category.



